Steady distillation, a form of distillation, is an ongoing separation by which a mixture is continuously (without interruption) fed into the method and separated fractions are eliminated repeatedly as output streams. Distillation is the separation or partial separation of a liquid feed mixture into components or fractions by selective boiling (or evaporation) and condensation. The process produces at the least two output fractions. These fractions include at least one risky distillate fraction, which has boiled and been separately captured as a vapor condensed to a liquid, and practically all the time a bottoms (or residuum) fraction, which is the least volatile residue that has not been individually captured as a condensed vapor.
An alternative to steady distillation is batch distillation, where the mixture is added to the unit in the beginning of the distillation, distillate fractions are taken out sequentially in time (one after one other) throughout the distillation, and the remaining bottoms fraction is eliminated at the top. As a result of each of the distillate fractions are taken out at different occasions, only one distillate exit point (location) is needed for a batch distillation and the distillate can simply be switched to a different receiver, a fraction-accumulating container. Batch distillation is usually used when smaller portions are distilled. In a continuous distillation, each of the fraction streams is taken concurrently all through operation; subsequently, a separate exit level is required for each fraction. In observe when there are a number of distillate fractions, the distillate exit points are located at completely different heights on a fractionating column. The bottoms fraction could be taken from the underside of the distillation column or unit, however is commonly taken from a reboiler linked to the underside of the column.
Each fraction may include a number of elements (forms of chemical compounds). When distilling crude oil or a similar feedstock, every fraction contains many parts of similar volatility and different properties. Though it is feasible to run a small-scale or laboratory continuous distillation, most often steady distillation is used in a large-scale industrial process.
1 Industrial utility
2 Precept
three Design and operation 3.1 Column feed
3.2 Bettering separation 3.2.1 Reflux
three.2.2 Plates or trays
3.2.Three Packing
4.1 Continuous distillation of crude oil
Industrial software[edit]
Distillation is likely one of the unit operations of chemical engineering.[1][2] Steady distillation is used broadly in the chemical process industries where giant portions of liquids have to be distilled.[Three][4][5] Such industries are the natural gas processing, petrochemical production, coal tar processing, liquor production, liquified air separation, hydrocarbon solvents manufacturing and similar industries, but it surely finds its widest application in petroleum refineries. In such refineries, the crude oil feedstock is a very complex multicomponent mixture that must be separated and yields of pure chemical compounds are usually not expected, solely teams of compounds inside a relatively small range of boiling points, that are known as fractions. These fractions are the origin of the term fractional distillation or fractionation. It is usually not worthwhile separating the elements in these fractions any additional based mostly on product requirements and economics.
Industrial distillation is typically carried out in large, vertical cylindrical columns (as shown in images 1 and a pair of) often known as “distillation towers” or “distillation columns” with diameters starting from about 65 centimeters to 11 meters and heights ranging from about 6 meters to 60 meters or more.
Principle[edit]
The precept for steady distillation is similar as for normal distillation: when a liquid mixture is heated so that it boils, the composition of the vapor above the liquid differs from the liquid composition. If this vapor is then separated and condensed right into a liquid, it turns into richer within the lower boiling point element(s) of the unique mixture.
That is what occurs in a steady distillation column. A mixture is heated up, and routed into the distillation column. On getting into the column, the feed starts flowing down but a part of it, the component(s) with lower boiling point(s), vaporizes and rises. Nevertheless, because it rises, it cools and whereas part of it continues up as vapor, a few of it (enriched in the less volatile part) begins to descend once more.
Picture three depicts a simple steady fractional distillation tower for separating a feed stream into two fractions, an overhead distillate product and a bottoms product. The “lightest” merchandise (these with the bottom boiling point or highest volatility) exit from the top of the columns and the “heaviest” products (the bottoms, those with the best boiling point) exit from the bottom of the column. The overhead stream could also be cooled and condensed using a water-cooled or air-cooled condenser. The bottoms reboiler could also be a steam-heated or scorching oil-heated heat exchanger, or perhaps a gas or oil-fired furnace.
In a steady distillation, the system is kept in a gradual state or approximate regular state. Steady state implies that quantities related to the method don’t change as time passes throughout operation. Such constant portions embody feed input charge, output stream rates, heating and cooling charges, reflux ratio, and temperatures, pressures, and compositions at each point (location). Unless the process is disturbed on account of adjustments in feed, heating, ambient temperature, or condensing, steady state is normally maintained. This is also the main attraction of continuous distillation, aside from the minimal quantity of (simply instrumentable) surveillance; if the feed price and feed composition are saved fixed, product price and high quality are additionally constant. Even when a variation in conditions happens, modern process control strategies are generally in a position to step by step return the steady process to another regular state once more.
Since a continuous distillation unit is fed continuously with a feed mixture and not filled abruptly like a batch distillation, a steady distillation unit does not want a sizable distillation pot, vessel, or reservoir for a batch fill. As an alternative, the mixture could be fed straight into the column, where the precise separation occurs. The height of the feed level along the column can range on the state of affairs and is designed so as to offer optimal results. See McCabe-Thiele methodology.
A continuous distillation is usually a fractional distillation and could be a vacuum distillation or a steam distillation.
Design and operation[edit]
Design and operation of a distillation column is determined by the feed and desired products. Given a simple, binary component feed, analytical methods such because the McCabe-Thiele technique[5][6][7] or the Fenske equation[5] can be utilized to assist in the design. For a multi-element feed, computerized simulation models are used both for design and subsequently in operation of the column as nicely. Modeling can be used to optimize already erected columns for the distillation of mixtures other than these the distillation tools was originally designed for.
When a steady distillation column is in operation, it needs to be closely monitored for changes in feed composition, operating temperature and product composition. Many of those duties are carried out utilizing advanced laptop control gear.
Column feed[edit]
The column will be fed in other ways. If the feed is from a supply at a strain greater than the distillation column strain, it is just piped into the column. In any other case, the feed is pumped or compressed into the column. The feed could also be a superheated vapor, a saturated vapor, a partially vaporized liquid-vapor mixture, a saturated liquid (i.e., liquid at its boiling point at the column’s stress), or a sub-cooled liquid. If the feed is a liquid at a a lot increased stress than the column stress and flows by way of a strain let-down valve just ahead of the column, it is going to instantly expand and bear a partial flash vaporization leading to a liquid-vapor mixture because it enters the distillation column.
Improving separation[edit]
Though small size models, largely fabricated from glass, can be used in laboratories, industrial items are massive, vertical, steel vessels (see images 1 and a couple of) often known as “distillation towers” or “distillation columns”. To improve the separation, the tower is generally supplied inside with horizontal plates or trays as proven in picture 5, or the column is full of a packing materials. To provide the heat required for the vaporization involved in distillation and also to compensate for heat loss, heat is most frequently added to the underside of the column by a reboiler, and the purity of the highest product might be improved by recycling a few of the externally condensed high product liquid as reflux. Relying on their purpose, distillation columns could have liquid outlets at intervals up the size of the column as shown in picture 4.
Reflux[edit]
Giant-scale industrial fractionation towers use reflux to attain more efficient separation of products.[3][5] Reflux refers back to the portion of the condensed overhead liquid product from a distillation tower that’s returned to the upper part of the tower as shown in pictures 3 and four. Contained in the tower, the downflowing reflux liquid gives cooling and partial condensation of the upflowing vapors, thereby increasing the efficacy of the distillation tower. The more reflux that is offered, the higher is the tower’s separation of the decrease boiling from the upper boiling components of the feed. A balance of heating with a reboiler at the underside of a column and cooling by condensed reflux at the top of the column maintains a temperature gradient (or gradual temperature distinction) along the top of the column to provide good situations for fractionating the feed mixture. Reflux flows at the center of the tower are called pumparounds.
Changing the reflux (together with modifications in feed and product withdrawal) may also be used to improve the separation properties of a continuous distillation column while in operation (in distinction to adding plates or trays, or altering the packing, which would, at a minimum, require fairly significant downtime).
Plates or trays[edit]
Distillation towers (similar to in photos 3 and 4) use various vapor and liquid contacting strategies to supply the required number of equilibrium levels. Such devices are commonly known as “plates” or “trays”.[8] Every of these plates or trays is at a different temperature and strain. The stage on the tower bottom has the highest strain and temperature. Progressing upwards in the tower, the strain and temperature decreases for every succeeding stage. The vapor-liquid equilibrium for each feed part within the tower reacts in its distinctive solution to the different pressure and temperature circumstances at every of the phases. Which means that every element establishes a unique concentration within the vapor and liquid phases at every of the phases, and this outcomes in the separation of the parts. Some instance trays are depicted in image 5. A extra detailed, expanded image of two trays could be seen in the theoretical plate article. The reboiler usually acts as an additional equilibrium stage.
If every physical tray or plate were 100% efficient, then the variety of physical trays needed for a given separation would equal the number of equilibrium levels or theoretical plates. Nevertheless, that may be very seldom the case. Therefore, a distillation column wants more plates than the required number of theoretical vapor-liquid equilibrium levels.
Packing[edit]
Another approach of bettering the separation in a distillation column is to make use of a packing material as a substitute of trays. These provide the benefit of a lower stress drop across the column (when compared to plates or trays), helpful when operating underneath vacuum. If a distillation tower uses packing as an alternative of trays, the number of mandatory theoretical equilibrium phases is first determined after which the packing top equal to a theoretical equilibrium stage, known as the peak equivalent to a theoretical plate (HETP), can be determined. The whole packing height required is the number of theoretical levels multiplied by the HETP.
This packing materials can both be random dumped packing comparable to Raschig rings or structured sheet metal. Liquids are likely to wet the surface of the packing and the vapors pass across this wetted surface, the place mass transfer takes place. Unlike typical tray distillation wherein every tray represents a separate level of vapor-liquid equilibrium, the vapor-liquid equilibrium curve in a packed column is steady. Nonetheless, when modeling packed columns it is helpful to compute a lot of theoretical plates to indicate the separation efficiency of the packed column with respect to extra traditional trays. Otherwise shaped packings have totally different surface areas and void house between packings. Both of those components have an effect on packing efficiency.
Another factor in addition to the packing form and floor space that impacts the performance of random or structured packing is liquid and vapor distribution getting into the packed mattress. The number of theoretical phases required to make a given separation is calculated using a particular vapor to liquid ratio. If the liquid and vapor should not evenly distributed across the superficial tower area because it enters the packed bed, the liquid to vapor ratio won’t be correct in the packed bed and the required separation is not going to be achieved. The packing will seem to not be working correctly. The top equal to a theoretical plate (HETP) will be larger than expected. The issue just isn’t the packing itself however the mal-distribution of the fluids coming into the packed mattress. Liquid mal-distribution is extra regularly the problem than vapor. The design of the liquid distributors used to introduce the feed and reflux to a packed bed is crucial to making the packing perform at maximum efficiency. Strategies of evaluating the effectiveness of a liquid distributor will be present in references.[9][10]
Overhead system arrangements[edit]
Photos four and 5 assume an overhead stream that is totally condensed right into a liquid product using water or air-cooling. However, in lots of cases, the tower overhead is just not simply condensed totally and the reflux drum should embrace a vent gas outlet stream. In but other circumstances, the overhead stream might also comprise water vapor as a result of either the feed stream comprises some water or some steam is injected into the distillation tower (which is the case in the crude oil distillation towers in oil refineries). In these cases, if the distillate product is insoluble in water, the reflux drum could comprise a condensed liquid distillate section, a condensed water part and a non-condensible gas phase, which makes it needed that the reflux drum also have a water outlet stream.
Examples[edit]
Continuous distillation of crude oil[edit]
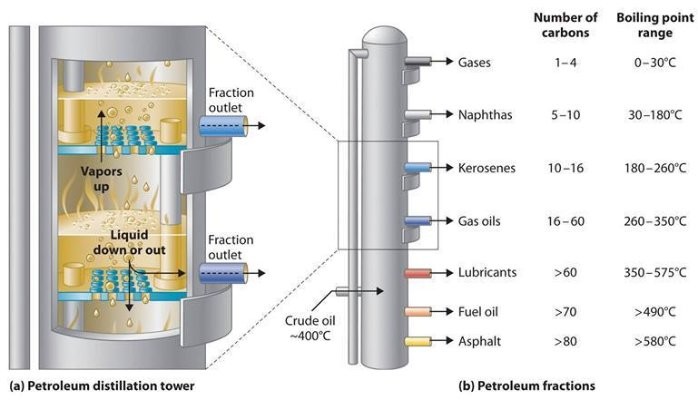
Petroleum crude oils comprise a whole lot of various hydrocarbon compounds: paraffins, naphthenes and aromatics as well as natural sulfur compounds, natural nitrogen compounds and a few oxygen containing hydrocarbons similar to phenols. Although crude oils usually don’t include olefins, they are formed in most of the processes utilized in a petroleum refinery.[11]
The crude oil fractionator doesn’t produce merchandise having a single boiling level; relatively, it produces fractions having boiling ranges.[11][12] For example, the crude oil fractionator produces an overhead fraction known as “naphtha” which turns into a gasoline element after it is additional processed through a catalytic hydrodesulfurizer to take away sulfur and a catalytic reformer to reform its hydrocarbon molecules into extra complex molecules with a better octane score value.
The naphtha cut, as that fraction is named, incorporates many alternative hydrocarbon compounds. Due to this fact, it has an preliminary boiling point of about 35 °C and a closing boiling point of about 200 °C. Every minimize produced within the fractionating columns has a unique boiling vary. At some distance beneath the overhead, the subsequent lower is withdrawn from the aspect of the column and it’s normally the jet gas minimize, also referred to as a kerosene cut. The boiling vary of that reduce is from an initial boiling level of about a hundred and fifty °C to a ultimate boiling level of about 270 °C, and it additionally incorporates many alternative hydrocarbons. The subsequent lower additional down the tower is the diesel oil lower with a boiling vary from about 180 °C to about 315 °C. The boiling ranges between any cut and the following minimize overlap because the distillation separations are usually not perfectly sharp. After these come the heavy gas oil cuts and at last the bottoms product, with very broad boiling ranges. All these cuts are processed further in subsequent refining processes.
Azeotropic distillation
Extractive distillation
Fractional distillation
Fractionating column
Steam distillation
^ Editors: Jacqueline I. Kroschwitz and Arza Seidel (2004). Kirk-Othmer Encyclopedia of Chemical Technology (fifth ed.). Hoboken, New Jersey: Wiley-Interscience. ISBN 0-471-48810-zero. CS1 maint: Additional text: authors listing (link)
^ McCabe, W., Smith, J. and Harriott, P. (2004). Unit Operations of Chemical Engineering (seventh ed.). McGraw Hill. ISBN 0-07-284823-5. CS1 maint: Multiple names: authors listing (hyperlink)
^ a b Kister, Henry Z. (1992). Distillation Design (1st ed.). McGraw-Hill. ISBN 0-07-034909-6.
^ King, C.J. (1980). Separation Processes (2nd ed.). McGraw Hill. ISBN 0-07-034612-7.
^ a b c d Perry, Robert H.; Inexperienced, Don W. (1984). Perry’s Chemical Engineers’ Handbook (sixth ed.). McGraw-Hill. ISBN zero-07-049479-7.
^ Beychok, Milton (Might 1951). “Algebraic Answer of McCabe-Thiele Diagram”. Chemical Engineering Progress.
^ Seader, J. D.; Henley, Ernest J. (1998). Separation Process Principles. New York: Wiley. ISBN zero-471-58626-9.
^ Pictures of bubble cap and other tray varieties (Webpage of Raschig Gmbh)
^ Random Packing, Vapor and Liquid Distribution: Liquid and gas distribution in industrial packed towers, Moore, F., Rukovena, F., Chemical Plants & Processing, Version Europe, August 1987, p. Eleven-15
^ Structured Packing, Liquid Distribution: A brand new methodology to evaluate liquid distributor high quality, Spiegel, L., Chemical Engineering and Processing 45 (2006), p. 1011-1017
^ a b Gary, J.H.; Handwerk, G.E. (1984). Petroleum Refining Technology and Economics (2nd ed.). Marcel Dekker, Inc. ISBN 0-8247-7150-8.
^ Nelson, W.L. (1958). Petroleum Refinery Engineering (4th ed.). McGraw Hill. LCCN 57010913.
Exterior hyperlinks[edit]
 Distillation Idea by Ivar J. Halvorsen and Sigurd Skogestad, Norwegian University of Science and Technology, Norway
Distillation Idea by Ivar J. Halvorsen and Sigurd Skogestad, Norwegian University of Science and Technology, Norway
Distillation by the Distillation Group, USA
Distillation Lecture Notes by Prof. Randall M.
If you have any concerns concerning where and how you can make use of petroleum equipment distributors, you could contact us at the site.